
Yesterday, I presented preliminary findings at the 2015 Clinical Genome Conference in San Francisco from our family trio sequencing project. In this crowdsourced project on experiment.com, I looked for genetic clues to autism in our adult-aged daughter. While the talk focused on the “DIY” aspects of how to accomplish WGS sequencing, this post focuses on genetic findings.
Overview
The project began with a crowdsourced effort to raise $1,750 to sequence our daughter’s genome, and took slightly more than two months to complete. After working with AllSeq and HudsonAlpha to obtain WGS data, we used VarSeq from Golden Helix to search for unique variants, as well as browse whole genome sequence data. After filtering our variant call data to focus on high quality exome variants, we examined 52 potentially damaging de novo and compound heterozygous changes suggested by VarSeq’s family trio analysis. Although this first approach did not yield clues specific to autism, it did suggest a number of secondary findings that are not addressed here. The second approach was to start with genes having known associations with autism and then look for them in our daughter’s DNA. Several curated databases have between 200 and 1200 genes, but again, none produced meaningful results. The third method was to look at known “hot spots” in autism genetics, such as variants in the NRXN1 gene, as well as known structural variation on chromosome 16. Changes to NRXN1 and so-called “16p” changes are discussed below.
Findings
 NRXN1 – Deletions in NRXN1 are associated with a wide spectrum of developmental disorders, including autism. Our daughter has a 10bp exonic deletion (-GT repeat) followed by what appears to be a 9bp compound heterozygous deletion in NRXN1. Both deletions are partially present in both parents and overlap; the deletions appear to have been accumulatively inherited. Due to the high number of sequence repeats, copy number variation (CNV) should clarify the significance of this finding.
NRXN1 – Deletions in NRXN1 are associated with a wide spectrum of developmental disorders, including autism. Our daughter has a 10bp exonic deletion (-GT repeat) followed by what appears to be a 9bp compound heterozygous deletion in NRXN1. Both deletions are partially present in both parents and overlap; the deletions appear to have been accumulatively inherited. Due to the high number of sequence repeats, copy number variation (CNV) should clarify the significance of this finding.

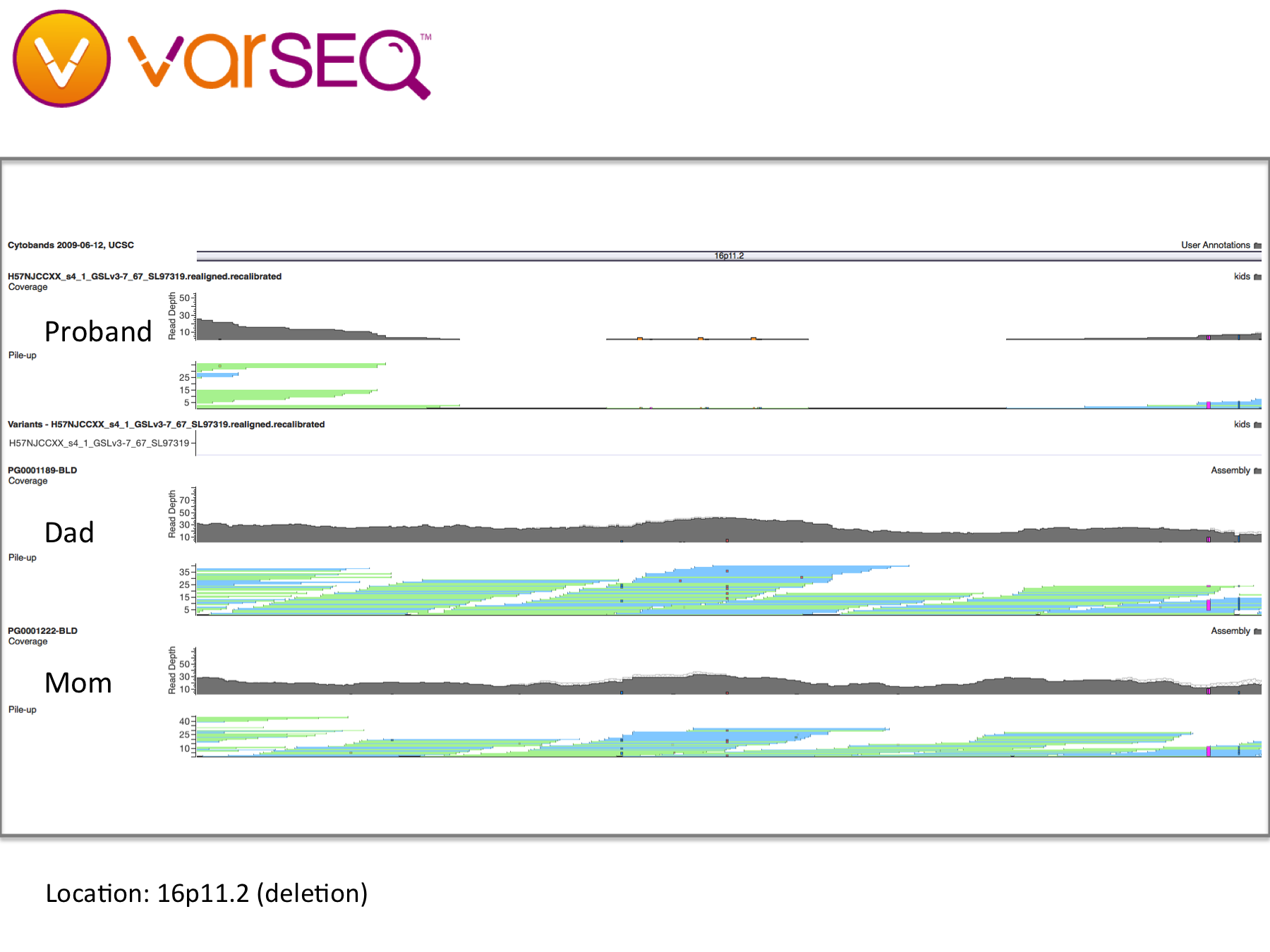
 16p deletions – Deletions and duplications in this 593-kilobase section of chromosome 16 are widely associated with developmental issues, including autism. Our daughter appears to have dozens of deletions in this region, some inherited and some not. However, since the variants in our daughter’s DNA were called using a different software pipeline, it is difficult to draw meaningful conclusions (see “Limitations,” below). For example, some variants in our daughter’s DNA were shown to map to multiple locations on the genome, suggesting either large copy number variation or genomic regions that were difficult to map. Copy number variation (CNV) analysis will also elucidate this region. Once reprocessed, these findings may provide potential genetic clues to our daughter’s condition.
16p deletions – Deletions and duplications in this 593-kilobase section of chromosome 16 are widely associated with developmental issues, including autism. Our daughter appears to have dozens of deletions in this region, some inherited and some not. However, since the variants in our daughter’s DNA were called using a different software pipeline, it is difficult to draw meaningful conclusions (see “Limitations,” below). For example, some variants in our daughter’s DNA were shown to map to multiple locations on the genome, suggesting either large copy number variation or genomic regions that were difficult to map. Copy number variation (CNV) analysis will also elucidate this region. Once reprocessed, these findings may provide potential genetic clues to our daughter’s condition.
Limitations
My wife and I received our WGS data in March 2014. Our samples were sequenced at 30x coverage using Illumina’s HiSeq platform and then aligned and called with Illumina’s pipeline, Isaac. Our daughter’s DNA was sequenced in May 2015 at 30x coverage, but on Illumina’s newest platform, the Illumina HiSeq X Ten. The difference is that our daughter’s DNA was aligned using BWA, followed by variant calling with GATK “best practice” workflow. To accurately compare genomes in family trio analysis, all samples must be processed using the same software pipeline. Otherwise, variants may be aligned and called differently. My wife and I must go back to the (almost) original FASTQ data and start over. Although Illumina did not provide these files with our results, Mike Lin explains how to extract FASTQ files from Illumina data in this great blog series. Hint: it involves a utility called Picard (no relation). Until we reprocess our WGS data using the same bioinformatics pipeline, all results should be considered preliminary.
Conclusion
This project demonstrated that personal genomics is very real, and has the potential to answer complex medical questions. Today, answering those questions using whole genome data and family trio analysis requires a combination of genetic, bioinformatic and domain knowledge to reach meaningful conclusions. Validating those conclusions remains challenging, from rare diseases to complex conditions such as autism. Currently, personal genomics has a similar feel to “homebrew” computer clubs from the late ’70s–the community is still very small, collegial, and willing to share “tips and tricks” to advance the field.
Although we encountered some “dark alleys” during the analysis, our preliminary results suggest that family trio sequencing can indeed provide genetic clues to autism. We will continue to refine the analysis by reprocessing the data with the same pipeline, which should resolve questions in the 16p region, as well as the potential deletion in NRXN1. Further, CNV analysis should answer structural variation questions that are also known to be associated with autism spectrum conditions.
Acknowledgements
I would like to thank our backers and the team at experiment.com, as well as Gabe Rudy from Golden Helix. Gabe was very generous with his time, knowledge and insight. Finally, I would like to thank my wife, Kimberly, for her patience and fortitude.
Additional resources: Slides
