This entry was cross-posted from Terra on April 28, 2021.
In April, we celebrate Citizen Science Month, World Autism Day, and National DNA Day. In this guest blog post, all three events come together as KT Pickard, father of a young woman with autism, shares his family’s story of personal genomics and citizen science.
This past Sunday was National DNA Day, which commemorates the discovery of DNA’s double helix in 1953 and the publication of the first draft of the human genome in 2003. Events on National DNA Day celebrate the latest genomic research and explore how those advances might impact our lives. Last year, I wrote a playful article for DNA Day that investigated whether genetics is truly like finding a needle in a haystack. This year, our family is honored to share our story and ideas with you.
Our family’s DNA odyssey
My wife and I have a young adult-aged daughter who is on the autism spectrum. We first discovered that our daughter had autism when she was eight years old. As we struggled to understand autism and what it meant for our family, we learned that autism is uniquely expressed: Meeting one person with autism means that you have met one person with autism.
Long fascinated with genomics, my wife and I wondered how our DNA may have contributed to her condition, and we set out to learn all that we could. It was the beginnings of this diagnostic odyssey that gave expression to my second career as a citizen scientist. My professional background in supercomputing, software engineering, and medical imaging were a good start to apply scientific principles and gain insights.
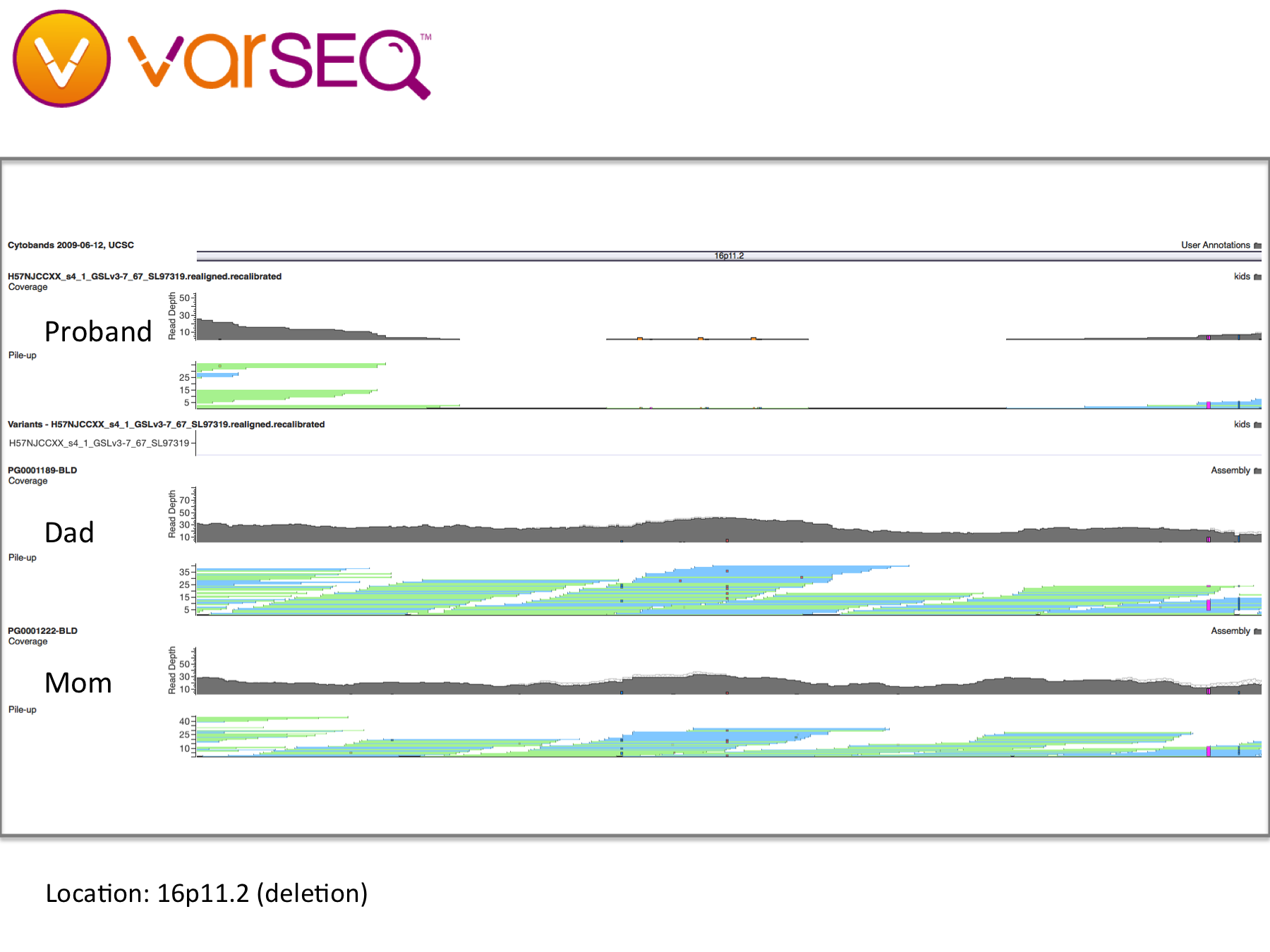
We began our journey by talking with our family doctor, then my wife and I had our whole genomes sequenced through the Understand Your Genome project. Later, we crowdsourced the sequencing of her genome and began looking for genetic clues. By applying trio analysis to our family data, we discovered some preliminary findings: Our daughter has deletions in the NRXN1 gene and in a large region of chromosome 16, which have been found to be widely associated with developmental issues including autism. It looks like my wife and I have each contributed some variant alleles, but we are being careful about interpreting these findings because our WGS data and our daughter’s were processed through different pipelines, which could lead to inconsistent results.

Trio analysis of the NRXN1 locus shows a compound heterozygous deletion, with each parent possibly contributing one allele (visualization by VarSeq from Golden Helix).
To continue our journey, I want to reprocess our family’s WGS data with the latest GATK Best Practices, in the hope that this will give us a consistent baseline. I came across Terra through the book Genomics in the Cloud, which I picked up to help me learn more about GATK. I led an online book club in early 2021 based on the book, and subsequently moved our WGS data to the Terra platform. Now I am using the GATK Whole Genome Analysis Pipeline in Terra to reprocess our data. Working with Terra has been challenging, but highly satisfying because it provides access to industry standard genomics tools.
From personal genomics to citizen science
My family’s main goal with this project is to make meaningful discoveries about the genetic basis of our daughter’s autism. In 2015, genetics could explain the heritability of autism spectrum disorder in approximately 1 in 5 cases. Amazingly, that number has increased to 4 in 5 cases today.

Yet there is more to be gained. Although whole genome sequencing may not provide directly actionable results for autism itself, WGS can make a huge difference for parents who discover a comorbid, but treatable condition. By sharing our data and our findings with others, we can accelerate medical knowledge.
A growing number of projects offer opportunities for non-scientists to contribute in various forms to the advancement of biomedical research. In U.S. healthcare, one of the largest citizen science projects—All of Us—seeks one million people to share their unique health data to speed up medical research. By creating a national resource that reflects and supports the broad diversity of the U.S., the goal of All of Us is to advance precision medicine for all.
We have enrolled in the All of Us project and are looking forward to doing our part. I find it inspiring that this is something we can all contribute to, as citizens, even those of us who are not researchers.
Looking to the future
At its core, citizen science is a collaboration between scientists and those who are curious and motivated to contribute to scientific knowledge. As our family’s odyssey unfolds, I like to reflect about what I see out here on the bleeding edge of research, and how it could be applied to improve outcomes for patients in the real-world.
In community practice, many medical providers have limited knowledge of autism. Due to a lack of effective data sharing and awareness, an undiagnosed person with autism who walks through the door of a hospital may appear like a rare disease patient. A clinician evaluating them would miss out on a huge amount of valuable context. How could we improve the system so that clinicians could more effectively recognize the underlying context of that person’s condition? We can address some of these issues with machine learning, but that requires pooling together huge amounts of data, and much of that data is difficult to access.
As a citizen scientist, I see an enormous opportunity to combine research data with real-world data and evidence across healthcare delivery organizations. Common ontologies and interoperability standards are making it increasingly easy to pool de-identified datasets to test hypotheses on synthetic data—realistic-but-not-real data—to gain insights. A recent “call to action” encourages citizen scientists to evaluate the utility of this method precisely because data can be shared without disclosing the identities of anyone involved. Done ethically and responsibly, this synthetic DNA approach has the potential to accelerate autism research and deliver new benefits to patients.
This is the perspective I have gained from my journey so far. By asking questions and continuing to discover more about what our genomes contain, I have been fortunate to learn much about scientific principles, bioinformatics, and a bit about the genetic basis of autism. Although it is at times a challenging road, I have found that the path of personal genomics and citizen science is a satisfying way to find answers to the questions that my family faces. I hope this story will inspire others to explore, and perhaps let researchers and clinicians see patients and their families as potential collaborators in the quest to understand complex conditions like autism.