Summary: My wife had breast cancer. These posts describe: 1) finding out, 2) genetic testing, 3) radiation therapy, and 4) an incidental finding in the APC gene.

Genetic testing
The day after Kimberly received her breast cancer diagnosis, we met with a board-certified genetic nurse, Frank. Before our visit, we completed a form that Frank used to create a family pedigree. The list below is not a pedigree, but it shows the history of cancer in Kimberly’s family. We removed kinship for privacy:
- Breast cancer (maternal side: 1, paternal side: 1)
- Lung/bone cancer (paternal: 3)
- Cervical cancer (maternal: 1)
- Colon cancer (paternal: 1)
- Liver cancer (paternal: 2)
- Kidney/bone cancer (paternal: 1)
- Esophageal cancer (paternal: 1)
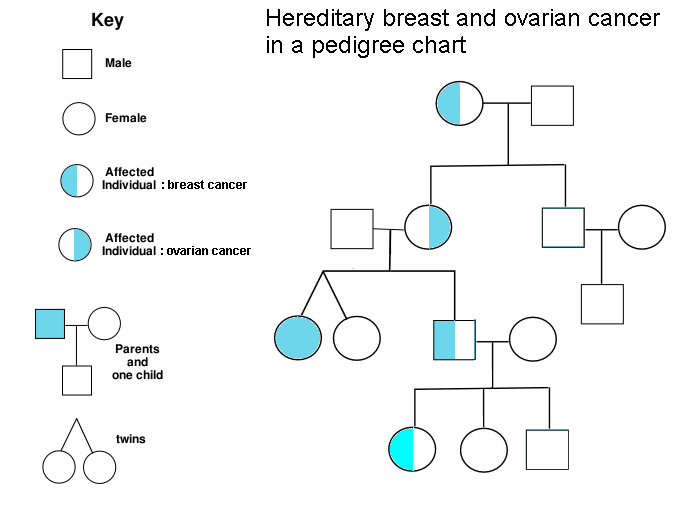
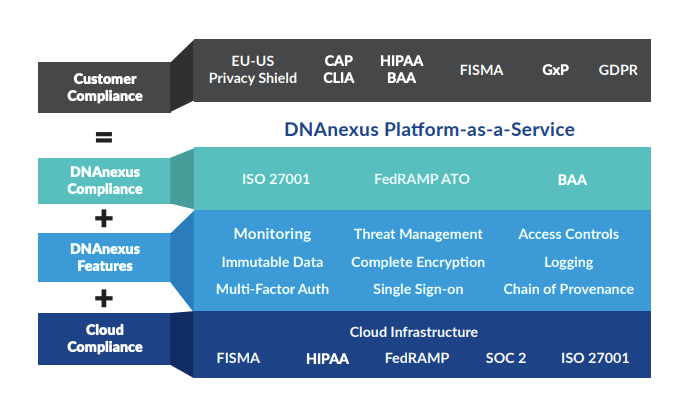
It turns out that 5-10% of cancer is inherited. People who carry hereditary mutations do not necessarily get cancer, but their lifetime risk is higher than average. Genetic counselors use pedigree charts to visualize family history and evaluate when genetic testing adds diagnostic value. Kimberly’s family history met lab guidelines for further evaluation, so Frank ordered a gene panel from a nearby lab, Invitae. The blood test was ordered stat, and we received our results six business days later. Treatment plans can change based on genetic results, so we were grateful to receive results before her surgery, which was now scheduled.
We returned to Frank’s office and first learned that she does not carry mutations in 9 genes known to influence the risk of breast cancer: ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, PTEN, STK11, and ΤΡ53. Phew! Invitae also provides free hereditary cancer testing to breast cancer patients at no additional charge (as long as you order the expanded panel within 90 days of the original test), so Frank ordered the expanded panel. Given that Kimberly has a family history of other hereditary cancers, we welcomed a broader genetic search. The results could be meaningful not only to us, but also to other living relatives. Oddly, our insurance company rejected all of our genetic testing claims because the resubmission was not related to her breast cancer diagnosis. I discussed the situation with Invitae and they were very accommodating–our total out-of-pocket cost was $250. I am still mad at our insurance company, but that’s a rant for another day.
Although she did not have any mutations related to breast cancer, Kimberly’s expanded genetic testing revealed a point mutation in the APC gene, which is known to increase the risk of colorectal cancer. People with this variant are generally counseled to have their first colonoscopy at age 40 (she did that) and follow-up colonoscopies every 5 years (coming up). Since the APC I1307K variant is autosomal dominant, close relatives such as siblings and children have a 1 in 2 chance of inheriting an APC mutation. We called Kimberly’s sister and shared our findings, part of a cascade testing strategy. We also have our kid’s whole genome sequences, which will let us check for APC mutations directly. We will return to that search in part 4 of this series.
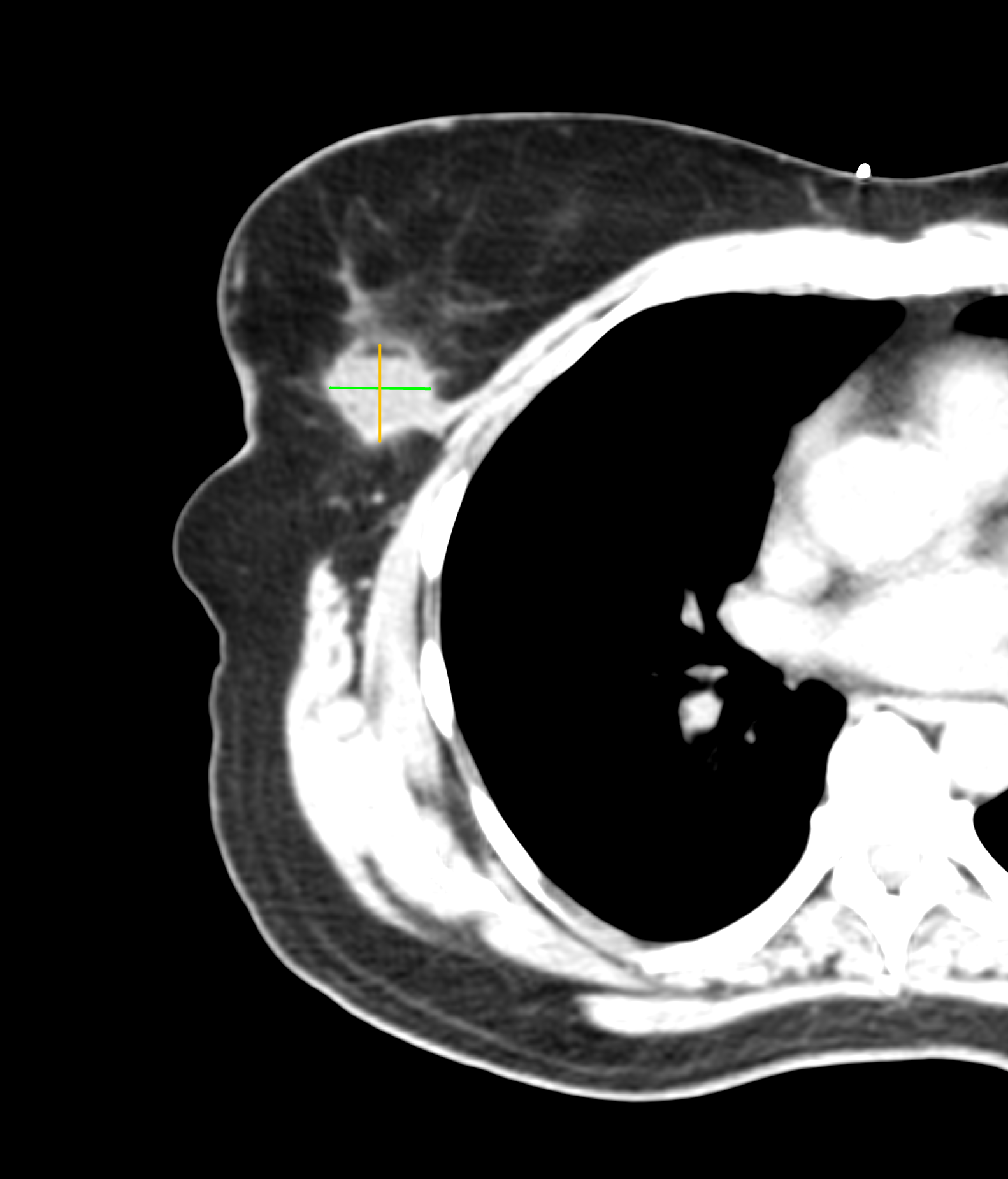
We left Frank’s office and developed a treatment plan with Kimberly’s surgeon and medical oncologist a few days later. The plan included surgery (lumpectomy) followed by radiation therapy. Surgery was successful, as you can see in the before and after images below. (Special thanks to the Horos Project for the open source DICOM viewer.)
Before surgery

After surgery
Kimberly received her diagnosis the day after Thanksgiving. In the 18 days that followed, we had 16 medical appointments that took us from diagnostic mammogram to surgery. With surgery behind us, Christmas was now six days away. We spent a quiet holiday with the kids.
We began 2020 hopeful, knowing that her type 1A tumor had been successfully removed by her surgeon. We were also much more knowledgeable about hereditary cancer risks due to Frank’s counseling.
One month later, Kimberly would begin radiation therapy to dramatically decrease her chance of recurrence.
/2